

Protein-DNA Interactions
Interactions between transcription factors (TFs) and their DNA binding sites are an integral part of regulatory networks within cells. These interactions control critical steps in progression through normal cellular processes and in responses to various environmental stresses. However, the DNA binding site specificities and regulatory functions of many known and most predicted DNA binding proteins are unknown. The Bulyk Lab has developed an improved in vitro protein binding microarray (PBM) technology to characterize TFs' sequence specificities in a high-throughput manner. The PBM technology allows us to determine the DNA binding site specificity of proteins in a single day. Comparison of binding site specificities determined from the PBM approach versus by in vivo genome-wide location analysis (ChIP-chip) indicates that the binding specificities can correspond well with each other. The universal PBM technology is depicted in the schematic below.
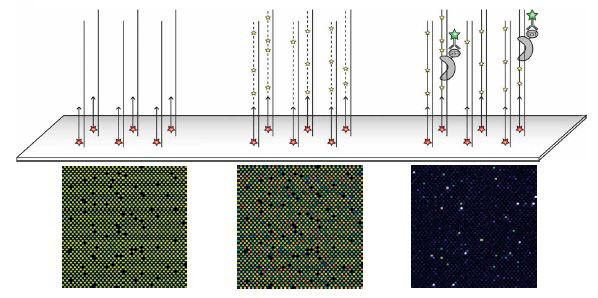
Universal PBM containing all possible 10-mer binding sites, bound by the S. cerevisiae TF Cbf1 expressed with a glutathione S-transferase (GST) epitope tag. Above is a schematic showing the three main stages of each experiment: primer annealing, primer extension, and protein binding. Beneath are zoom-in images of each stage for the same microarray, scanned at different wavelengths: Cy5-labeled universal primer, Cy3-labeled dUTP and Alexa488-conjugated a-GST antibody. Fluorescence intensities are shown in false color, with blue indicating low signal intensity, green indicating moderate signal intensity, yellow indicating high signal intensity, and white indicating saturated signal intensity. The variability observed in the Cy3-dUTP signal is due to differences in the nucleotide composition of each feature. The blank spots are single-stranded negative control probes that do not contain the universal primer sequence.
Universal Protein Binding Microarrays
 In order to analyze transcription factors from other species, we created a compact, universal PBM that contains all possible sequence variants of a given length k (i.e., all k-mers). Our design is based on de Bruijn sequences, in which all k-mers can be represented in an overlapping manner in an unbiased fashion. To date we have constructed such 'all k-mer' PBMs covering all 10 bp binding sites using high-density Agilent microarrays, enabling us to fit all 1,048,576 10-mers in approximately 44,000 spots. Using these microarrays, we comprehensively determined the binding specificities over a full range of affinities of >500 TFs from a wide range of structural classes and organisms. Our universal PBMs permit the discovery of subtle preferences in transcription factor binding sites (including interdependencies among different positions) and can be used with transcription factors from any species regardless of the level to which its genome has been characterized. We have also examined human and mouse NF-kB dimers using a custom-designed NF-kB PBM that highlighted dimer-specific differences.
In order to analyze transcription factors from other species, we created a compact, universal PBM that contains all possible sequence variants of a given length k (i.e., all k-mers). Our design is based on de Bruijn sequences, in which all k-mers can be represented in an overlapping manner in an unbiased fashion. To date we have constructed such 'all k-mer' PBMs covering all 10 bp binding sites using high-density Agilent microarrays, enabling us to fit all 1,048,576 10-mers in approximately 44,000 spots. Using these microarrays, we comprehensively determined the binding specificities over a full range of affinities of >500 TFs from a wide range of structural classes and organisms. Our universal PBMs permit the discovery of subtle preferences in transcription factor binding sites (including interdependencies among different positions) and can be used with transcription factors from any species regardless of the level to which its genome has been characterized. We have also examined human and mouse NF-kB dimers using a custom-designed NF-kB PBM that highlighted dimer-specific differences.
SELEX-Seq
 Alternatively, one can instead employ in vitro selection and sequencing protocols (e.g., SELEX-Seq) to assay TF DNA binding specificity. Briefly, a TF of interest is incubated with a double-stranded DNA library. Multiple rounds of selection are performed to capture DNA sequences bound specifically by the TF. After each round of selection, the selected ligand is PCR-amplified and purified for use as ligand in the next round. The selected DNAs from each of round of selection, plus an aliquot of the starting library, are sequenced and then analyzed to determine the DNA binding specificity of the TF.
Alternatively, one can instead employ in vitro selection and sequencing protocols (e.g., SELEX-Seq) to assay TF DNA binding specificity. Briefly, a TF of interest is incubated with a double-stranded DNA library. Multiple rounds of selection are performed to capture DNA sequences bound specifically by the TF. After each round of selection, the selected ligand is PCR-amplified and purified for use as ligand in the next round. The selected DNAs from each of round of selection, plus an aliquot of the starting library, are sequenced and then analyzed to determine the DNA binding specificity of the TF.